

Introduction to Raman Spectroscopy
Raman scattering (or the Raman effect) was discovered in 1928 by V. C. Raman who won the
Nobel prize for his work. If the substance being studied is illuminated by monochromatic light, for example from a laser, the spectrum of the scattered light consists of a strong line (the exciting line) of the same frequency as the incident illumination together with weaker lines on either side shifted from the strong line by frequencies ranging from a few to about 3500 cm-1. The lines of frequency less than the exciting lines are called Stokes lines, the others anti-Stokes lines.
Raman spectroscopy is very important practical tool for quickly identifying molecules and minerals.
A Raman spectrometer was deployed on the Viking landers in 1972 and in other missions.
Raman spectroscopy also has important scientific applications in studying molecular structure.
Raman Effect & Raman Scattering
When light is scattered from a molecule or crystal, most photons are elastically scattered. The scattered photons have the same energy (frequency) and, therefore, wavelength, as the incident photons.
However, a small fraction of light (approximately 1 in 107 photons) is scattered at optical frequencies different from, and usually lower than, the frequency of the incident photons.
The process leading to this inelastic scatter is termed the Raman effect. Raman scattering can occur with a change in vibrational, rotational or electronic energy of a molecule. If the scattering is elastic, the process is called Rayleigh scattering. If it’s not elastic, the process is called Raman scattering.
The Raman effect arises when a photon is incident on a molecule and interacts with the electric dipole of the molecule. In quantum mechanics the scattering is described as an excitation to a virtual state lower in energy than a real electronic transition with nearly coincident de-excitation and a change in vibrational energy. The scattering event occurs in 10-14 seconds or less. The virtual state description of the scattering is shown in Figure 1.
The energy difference between the incident and scattered photons is represented by the arrows of different lengths in Figure 1.
Numerically, the Raman shift in wave numbers (cm^-1), is calculated
through Eq. 1,
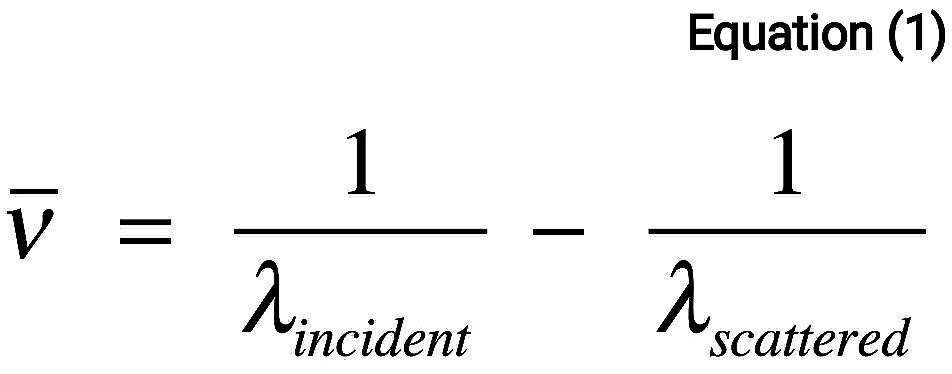
in which the λ’s are the wavelengths (in cm) of the incident and Raman scattered photons, respectively

At room temperature the thermal population of vibrational excited states is low, although not zero.
Therefore, the initial state is the ground state, and the scattered photon will have lower energy (longer wavelength) than the exciting photon. This Stokes shifted scatter is what is usually observed in Raman spectroscopy. Figure 1(a) depicts Raman Stokes scattering.
A small fraction of the molecules are in vibrationally excited states. Raman scattering from vibrationally excited molecules leaves the molecule in the ground state. The scattered photon appears at higher energy, as shown in Figure 1(b). This anti-Stokes-shifted Raman spectrum is always weaker than the Stokes-shifted spectrum, but at room temperature it is strong enough to be useful for vibrational frequencies less than about 1500 cm-1.
The Stokes and anti-Stokes spectra contain the same frequency information. The Anti-Stokes spectrum can be used when the Stokes spectrum is not directly observable, for example, because of poor detector response at lower frequencies.